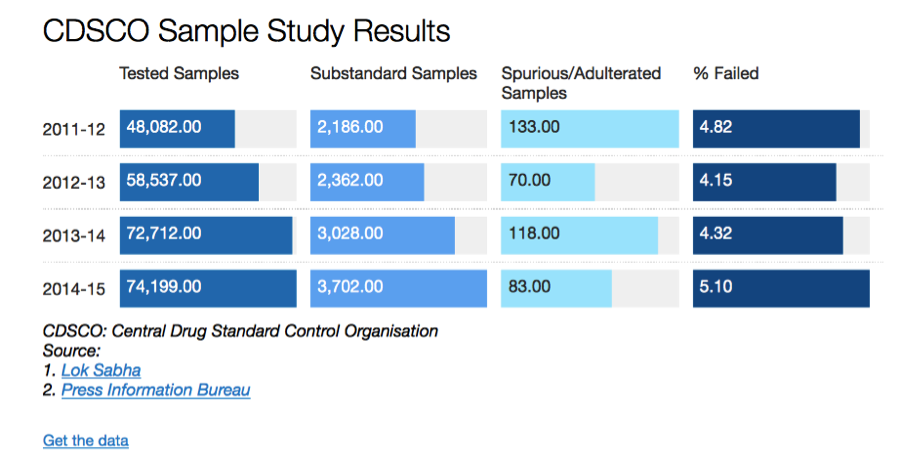
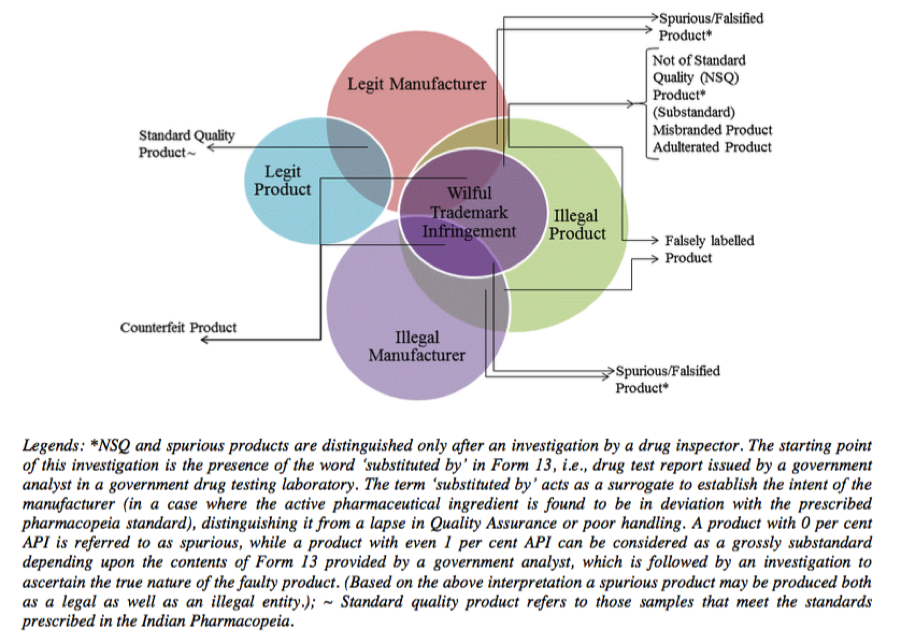
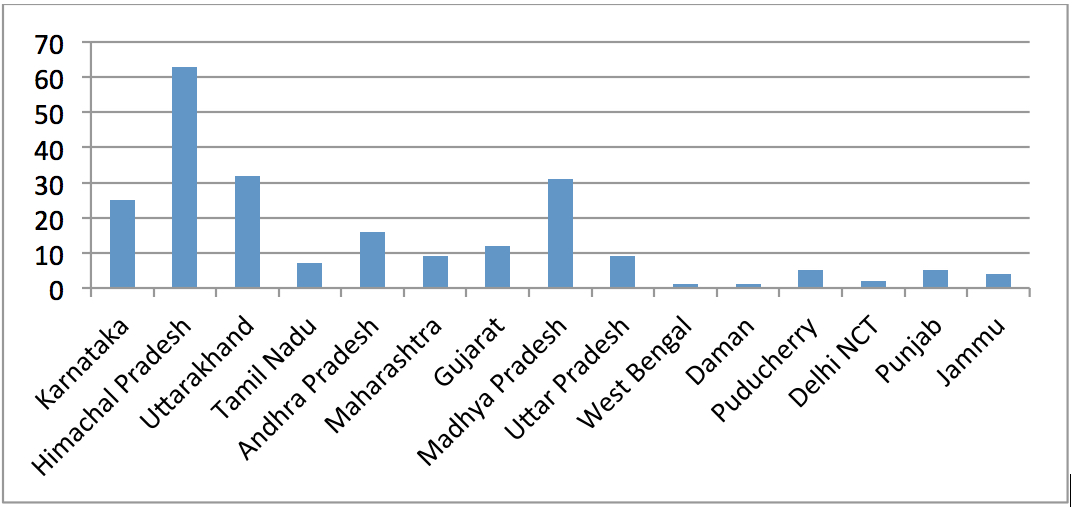
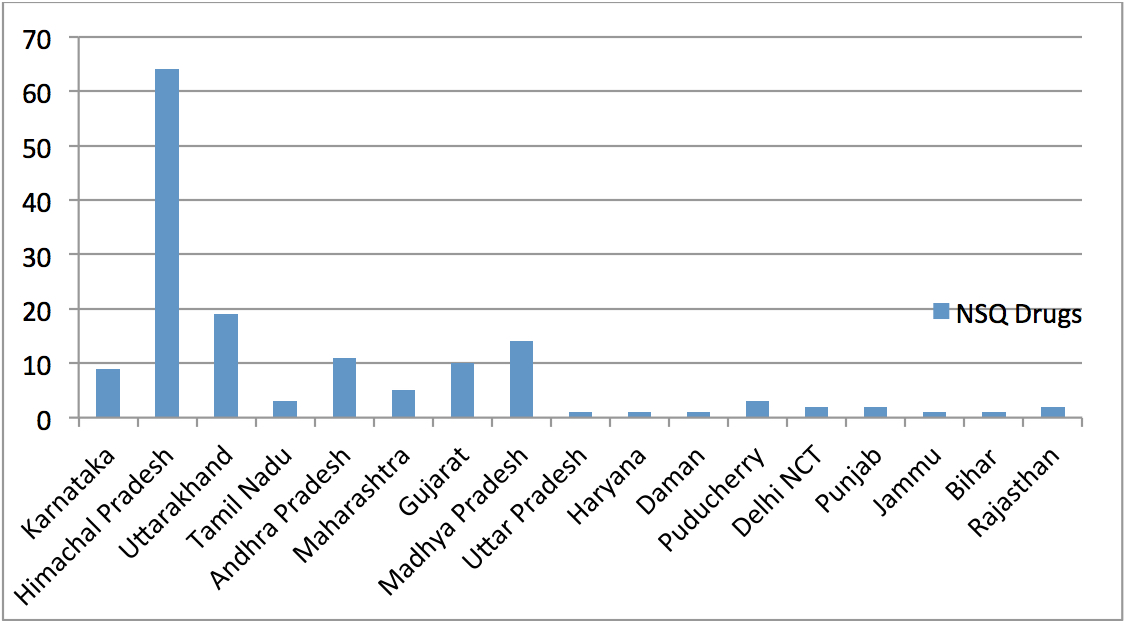
Over the last several years, the Comptroller & Auditor General (CAG) has pointed out to serious deficiencies in the public procurement of medicine by major institutions like the Ministry of Railways, Ministry of Defence and the Central Government Health Scheme (CGHS), which is administered by the Ministry of Health & Family Welfare. Some of the findings of these various audit reports are highlighted below:
For samples which were tested from the Armed Forces Medical Stores Depot (AMFS), the CAG report notes that the rate of rejection for locally procured medicine, due to samples failing quality tests, increased from 15% to 31% during 2006-07 to 2010-11. The average rate of rejection during the three year period of 2008-09 to 2010-11 was therefore 24% approximately. This means that one in every four drugs dispensed by these organizations is not of standard quality. This is a shockingly high rate of NSQ drugs which are used to treat people who fight for our country.
In its report no. 28 of 2014 on the Railways Hospitals, the CAG noted that substandard drugs worth Rs. 21.45 lakh were supplied to 20 hospitals over 8 different zones of the railways. The actual figure is most likely higher because as also noted in the same CAG report, the railways hospitals were not conducting mandated pre-dispensing testing of consignments i.e., each consignment of these drugs is required to be tested before being issued to patients. Such pre-dispensing testing is a testimony to the lack of faith in the ability of the DCGI and state drug controllers to effectively regulate quality standards. Can you imagine a drug approved and regulated by the US FDA being subject to this kind of procedure?
Even in cases where pre-dispensing quality testing is conducted, it was found that in 8 hospitals, over 4 railways zones, had dispensed these drugs to patients and then received the test reports indicating that the drugs were NSQ. In one case in Kolkata, 93.8% of a batch of drugs were dispensed before the test reports returned from the lab. Imagine what consequences the patients who got these drugs suffered. In most of these cases, CAG noted that information regarding these suppliers of NSQ drugs was not shared on railnet, “an internal portal”, for information to other zones. Independent of the CAG report, we also filed a RTI application with the Ministry of Railways asking for the names of all the pharmaceutical companies blacklisted by the Indian Railways. To our surprise, we learnt that the Indian Railways does not have a single consolidated blacklist of all pharmaceutical companies which have been debarred from supplying to the Railways because of poor quality products that they supply. Instead our RTI application was transferred to each zonal railways office. We found that each zone had its own blacklist thereby giving rise to the probability that a supplier blacklisted by one zone can still supply to other zone. Of all the zones which provided replies, only the Western Railways, NorthWestern Railways, North-East Frontier Railways & Eastern Zone even had a blacklist. Some of the companies on the list were rather big names like Biocon (blacklisted for Rosuvastatin), RPG LifeSciences (blacklisted for Atorvastatin), Sandoz, Alkem, Alembic Pharmaceuticals, Abbot etc. Some manufacturers like Ind-Swift & CMG Biotech Pvt. Ltd. were blacklisted for all of their products, while the others were blacklisted for only specific drugs that they supplied. Most of the other zones like the Southern Railways, Northern Railways, South Central Railways, East Coast Railways all claimed that they had not blacklisted evena single manufacturer. The lack of a consolidated blacklist is likely creating windows of opportunity for the manufacturers of sub-standard drugs to supply to one zone even after being blacklisted by others.
The CAG Report No. 20 of 2007 (Performance Audit) titled ‘Procurement of Medicines and Medical Equipment’ by government hospitals and CGHS pointed out several deficiencies in the procurement process. Essentially, the entire procurement process was punctuated by completely arbitrary behaviour and lack of set processes or guidelines. One of the key deficiencies pointed out with regard to quality control was the failure of hospitals, including AIIMS to carry out mandatory testing on all procurements before issuing. (para 7.1.8) The CAG report spurred a more detailed examination of the CGHS processes by the Public Accounts Committee (PAC) of the Lok Sabha in its 24th Report (2011) and 84th Report (2013). Some changes were made by the MoHFW, but clearly the changes were not enough because as noted by the PAC in its 22nd Report (2015), sub-standard drugs in the CGHS were still a problem. As noted by the Committee in its report between 2009-2012, CGHS, Bombay had reported Rs. 28.45 lakhs worth of drugs as sub-standard. Of these medicines, stock worth Rs. 15.66 lakhs had already been issued to patients. The Committee had noted “Such instances highlight the absence of a robust mechanism for quality assurance, which exposes the patients to the hazards of sub-standard medicines and drugs”.
In the backdrop of the above reports, we attempted to scrutinise the blacklisting policies followed by the above public authorities to penalise the suppliers of sub-standard drugs. Such blacklisting policies are important because very often, blacklisting is the only punishment meted out to suppliers of sub-standard drugs. Prosecutions are very rare in the Indian context as I have documented in my previous blogs.
As of now, each Ministry follows its own procurement and blacklisting guidelines. While individual Ministries may have different needs, it is surprising to see the degree of variability in each of their blacklisting guidelines. Here are some obvious holes in their procedures:
- The CGHS blacklisting guidelines are contained in the “Procurement and Operational Manual for Medical Store Organisation and Government Medical Store Depots”. The guidelines basically borrow the classification of various defects with sub-standard medicine from certain DCC Guidelines which creates a classification mechanism of Category A, Category B & Category C defects. In the context of the procurement manual, Category A defect in a product results in the supplier being barred for 3 years and if there is a repeat, then the supplier is barred from supplying any products. Category B defects are treated similarly. Such a system of blacklisting is however rather superficial and fails to understand the nature of the pharmaceutical industry. If a particular batch of medicine fails quality control testing at a certified GMP manufacturing facility (as all Indian pharmaceutical facilities are required to be), it would mean that the facility is not GMP compliant because by their very nature, GMPs create a fool proof mechanism to ensure quality. Every batch has to be tested before it is shipped and the manufacturer has to test the samples before shipping out commercial supplies. In many cases in India, manufacturing problems arise due to non-compliance with GMPs and the defects within a particular batch are merely a symptom of a larger problem within the company. This is the reason why we see a string of warning letters from foreign regulators to the who’s-who of the Indian pharmaceutical industry. Therefore, when a public authority detects quality issues with a particular batch of drugs, it should conduct a deeper investigation and determine the reasons for the problem – in some cases it could be purely a case of cheating or fraud by the supplier to make more profits. In such a case, the entire manufacturing facility should be banned, there is no point of banning the supplier only the one product which has failed the quality control test.
- MoD’s Policy Regarding Quality Assurance of Drugs and Punitive Action: This policy laid down by the Directorate of Quality Assurance (Stores) follows the same logic as the CGHS guidelines although the parameters for classifying defects are entirely different.
- The Indian Railways has its own Drug Procurement Policy, 2014. Unlike the MoD or the CGHS guidelines, the Railways doesn’t lay down a product specific blacklisting policy. The guidelines states that if there are adverse reports regarding the performance of a firm, the railways officers will inspect the facility and if the firm continues to fail to comply with orders of the Railway to improve quality it will be deregistered. What makes the Railways officials expert at detecting compliance problems at the manufacturer when they have no formal training in conducting such inspections is a question that is never asked. In practice however, from the blacklists provided to us by the Railways, it is quite clear that some zones like the Western Railways are actually following a product wise ban which is contrary to what is stated in the policy.
Apart from an urgent need to completely replace these blacklisting norms, another major reform required to the current process is to create a consolidated blacklist for all publicly funded entities. As noted in the CAG report, even within the Railways there is little sharing of information on NSQ suppliers.
Given the large amounts of money spent on public procurement of drugs, stronger quality controls norms will not only save public funds but also raise the costs for the inefficient firms who aren’t able to maintain quality. More importantly, they will close the loopholes that the drug companies use to exploit the system.
The best way to achieve the above objectives is for Parliament to enact a law dealing with public procurement of medicine. This was one of the prayers that I had sought in my PIL. Unfortunately, the Supreme Court thought that this was an academic issue.